Pancuronium For Anaesthetists
22 March 2026
Contents
Introduction
Pancuronium bromide is the original aminosteroid neuromuscular blocking agent arriving in clinical practice in the late 1960s and paved the way for every aminosteroid that followed. While it has been superseded by rocuronium and vecuronium in modern anaesthetic practice, understanding pancuronium’s pharmacology gives you a solid foundation for the aminosteroid class as a whole; it sits at the margin of the FRCA Primary curriculum but underpins the aminosteroid class.
We then take a detailed look at Type I and Type II neuromuscular blockade: what happens at the receptor level when suxamethonium is given once versus repeatedly, how we might think Phase II block mimics a non-depolarising pattern on the nerve stimulator, and how anticholinesterases interact differently with each block type.
Pancuronium Pharmacology
Pancuronium is a long-acting, bis-quaternary aminosteroid that competitively antagonises acetylcholine at the nicotinic N₂ receptor of the neuromuscular junction.
Pancuronium Physico-Chemical Properties
| Name | Pancuronium Bromide ‘Pavulon’ Derived: The name was derived from p(iperidino)an(drostane)cur(arising)-onium. |
| Class | Bis-Quarternary Aminosteroid |
| Chemical Make Up | Whats the craic with Aminosteroids? The gist of the situation is, they plonked a acetylcholine like structure on a 5-Alpha-androstane-17-one / 5-alpha-prenan20-one – the former a synthetic androstenedione then later an endogenous steroid hormone and found that it had a bit of paralysing activity, and the race was on to develop something useful! – A paper from Buckett et Al reports: ‘Although of limited clinical value, such compounds present useful leads for further chemical exploration, particularly the synthesis of bisquaternary amino-steroidal salts.’ A series of 2,8,16,3-diamino-5a-androstane-3a,17p-diol dimethohalide derivatives was described by Buckett, Hewett & Savage (1967). One of them, 2ft,16,/-dipiperidino-5a-androstane- 3a,17,8-diol diacetate dimethobromide (code number Org.NA97; approved name pancuronium bromide. |
| History | Synthesised in 1964 by Hewett and Savage A side note, to ‘Bowmans Principle’ of onset being inversely proportional to potency, coined by a behemoth of a boffin, Prof Bill Bowman, sadly not of this world now but spent his university of strathclyde career absolutely bossing neuromuscular pharmacology. |
| Isomer Status | Tis a single stereoisomer |
| Colour/Appearance | Odourless white crystalline powder (with a bitter, astringent taste!) pH 4 Unstable in Air 4mg in an ampoule |
| Molecular weight | 732 g/mol [mivacurium](/episode/mivacurium/)= 1029.3 g/mol, Roc 529, suxamethonium 361, trac 929) |
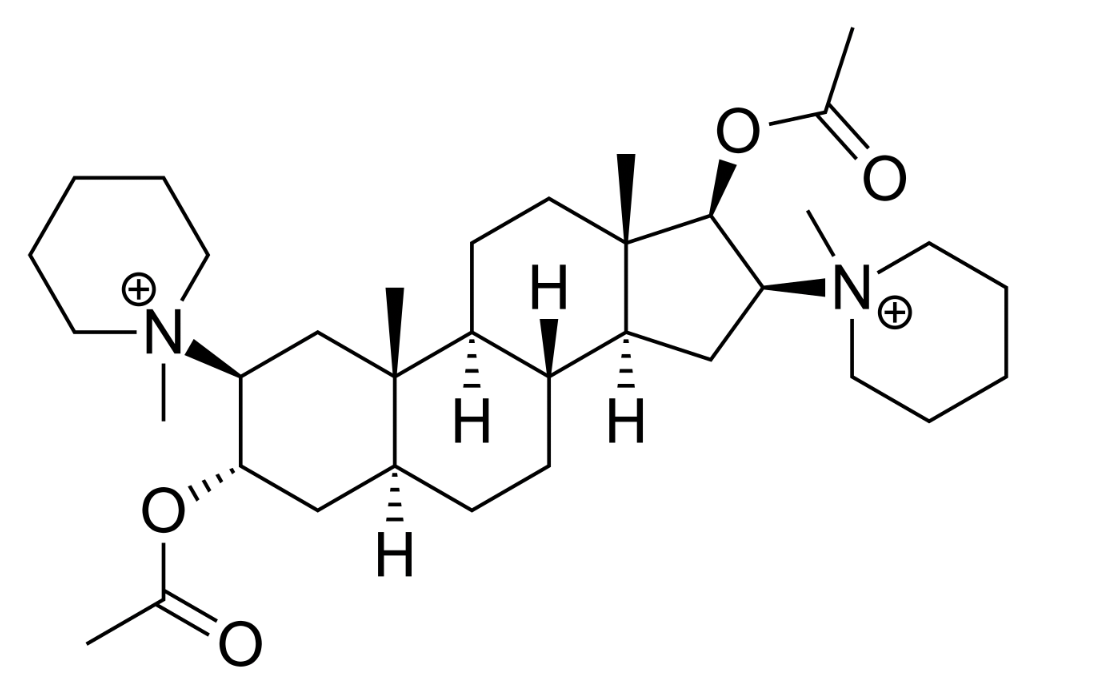
{kind=link}
A Pancuronium molecule!
Pancuronium Pharmacodynamics & Side Effects
| Mechanism of Action | Competitive non-depolarising neuromuscular blockade (Check out rocuronium for more details) |
| Chief Effect / Actions | Antagonism of acetylcholine at the nicotinic (N2) receptor situated on the post synaptic membrane of the skeletal neuromuscular junction. Does have a ‘slight; action in the ganglion blocking department but when compared against other agents at the time, was considered a much better choice! |
| Dose | Intubating – 0.1mg / kg – IV – Onset :~2.5+ mins Maintenance – Not often needed as it lasts for yonks Lowest paralysing dose in non-human model is 0.01mg/kg Caution, long acting relaxants, will have a longer time between being ‘reversible’ and not needing reversal. We care because if the neostigmine offsets before the relaxant has actually worn off, you could end up with a floppy patient (who might be on the ward, and on the post surgical obs regimen which whilst initially they are closely observed, does stretch out……) Onset time – faster that tubocurarine / Gallamine, lasting longer than gallamine, and similarly lenmgth of effect as tubocurarine Offset – at least 90 minutes ORAL LD50! in mice! a mouse must have 21.9 mg/kg of pancuronium orally to be rendered paralysed and thus doomed. So one would take a stab that rocuronium is possibly orally absorbed. but to achieve a lethal oral dose would take significant effort with 21×75 = ~1500mg of pancuronium to cause a problem, so thats 400 ampoules…. if we were to consider roc and panc equipotent, that would be 31x50mg ampoules of rocuronium! but pan is perhaps 3-4x the potency of rocuronium., so perhaps 120 ampoules of rocuronium….. |
| Cardio-Vascular Side Effects | Headline : Increase HR/BP and CO secondary to vagolytic effect. Mechanism: muscarinic M2 receptor blockade at the sinoatrial node. Histamine release very unlikely. |
| Respiratory Side Effects | Rate – Obliterated Depth – Obliterated Parenchymal effects: nil |
| Metabolic/MSK: Side Effects | Effect extended by volatile anaesthetic agents / gentamicin / magnesium etc (mechanism: reduced presynaptic ACh release with aminoglycosides; Ca²⁺ antagonism with magnesium; reduced post-junctional sensitivity with volatiles) |
Pancuronium Pharmacokinetics
| Absorption | n/a |
| Distribution | Volume of distribution 0.2 L/kg (but higher in cirrhotic patients, potentially necessitating a higher dose.) Big, polar molecule, doesn’t exactly float off into adipose tissue. |
| Metabolism Remember: Phase I : [oxidation, reduction, hydrolysis] (more cytochrome action here) (more O2 needed) occurs in inner aspect of liver acinus… Phase II: [conjugation, glucoronidation, acetylation, sulphylation] (less O2 needed) occurs in outer aspect of liver acinus… | 30-45% undergoes hepatic metabolism by deacetylation, >biliary excretion a metabolite – 3-hydroxy-pancuronium (1/4 of metabolic product has 50% the activity of pancuronium. |
| Elimination | Half-life elimination Halflife of 69-161 mins. – This is ~doubled if you’ve a cirrhotic patient. Clearance 1.1-2.2 ml/kg/min 40-50% excreted in the urine. (80% of this is unchanged drug) |
Type I and Type II Neuromuscular Blockade
Type I (depolarising) block is caused by suxamethonium at standard doses; Type II (desensitisation) block develops with cumulative doses and mimics non-depolarising blockade on the nerve stimulator.
There are two patterns of neuromuscular blockade Type I (depolarising) and Type II (desensitisation/dual block) both caused by Suxamethonium. We shall be exploring what is known about this phenomenon in a little more detail.
TOF in a suxamethonium paralysed patient? We don’t quite think about this, as who twitches a post suxamethonium patient?
Suxamethonium, a poison chalice
Side effects are legion, but those fasciculation, and the onset time! (just give a decent dose i.e. somewhere between 1-2 mg/kg)
- Malignant hyperthermia.
- Suxamethonium apnoea.
- Anaphylaxis.
- Significant bronchospasm.
- Myalgia.
- Life-threatening hyperkalaemia in patients with denervation injuries, muscular dystrophy, significant cerebral palsy, or big burns whicha re >48 hours old (due to extra-junctional nicotinic receptor upregulation).
- Raised intraocular pressure and intracranial pressure.
- Bradycardia with repeat doses (especially in children).
- Phase II block in repeat dose.
Type I Block
- Suxamethonium acts as an agonist at post-junctional N₂ receptors, causing depolarisation.
- Fasciculations arise from disordered depolarisation across the muscle as suxamethonium reaches different motor endplates at slightly different times.
- Acetylcholinesterase in the neuromuscular junction cannot metabolise suxamethonium; it must diffuse back to plasma for clearance by plasma cholinesterase (butyrylcholinesterase).
- No fade on train-of-four: each stimulus produces the same diminished amplitude because suxamethonium binding is non-competitive.
- No post-tetanic potentiation.
Type II Block
- Resembles a non-depolarising block despite being caused by a depolarising drug.
- Demonstrates fade on train-of-four, tetanic fade, and post-tetanic potentiation.
- The nicotinic receptor transitions from an open (depolarised) state to a desensitised (closed) state with suxamethonium still bound.
- Na⁺/K⁺-ATPases gradually restore membrane potential, but closed receptors remain unresponsive.
- Standard dose (1–2 mg/kg) remains in Phase I; repeated/cumulative doses (3–5 mg/kg) shift to Phase II.
- Tachyphylaxis develops: progressively more suxamethonium needed.
- Giving rocuronium to a patient in Phase II block deepens paralysis.
- Anticholinesterases may partially reverse a Phase II block (increased acetylcholine outcompetes at open receptors).
- Neostigmine given during a Phase I block prolongs the depolarising blockade.
Summary
Phase I block is a straightforward depolarisation block, no fade, no post-tetanic potentiation, reversed by drug leaving the NMJ. Phase II block develops with prolonged or repeated suxamethonium exposure and mimics competitive blockade on monitoring with fade, post-tetanic potentiation, and partial reversibility with anticholinesterases. The mechanism is incompletely understood but involves receptor desensitisation and possibly presynaptic mischief. Depolarising and non-depolarising agents interact at the receptor level. The transition from Phase I to Phase II is dose-dependent, relatively abrupt, and coincides with tachyphylaxis.
References
- Dobkin, A.B., Evers, W., Ghanooni, S. et al. Pancuronium bromide (Pavulon®) evaluation of its clinical pharmacology. Canad. Anaesth. Soc. J. 18, 512–535 (1971). https://doi.org/10.1007/BF03026014
- BUCKETT, W.R., MARJORIBANKS, C.E.B., MARWICK, F.A. and MORTON, M.B. (1968), THE PHARMACOLOGY OF PANCURONIUM BROMIDE (ORG.NA97), A NEW POTENT STEROIDAL NEUROMUSCULAR BLOCKING AGENT. British Journal of Pharmacology and Chemotherapy, 32: 671-682. https://doi.org/10.1111/j.1476-5381.1968.tb00466.x
- McKenzie, A.G. (2000), Prelude to pancuronium and vecuronium. Anaesthesia, 55: 551-556. https://doi.org/10.1046/j.1365-2044.2000.01423.x
- Appiah-Ankam J, Hunter J Pharmacology of neuromuscular blocking drugs Continuing Education in Anaesthesia, Critical Care and Pain, 4, 2-7
- Lee C. Goodbye suxamethonium! Anaesthesia 2009; 64(Suppl 1): 73–81.
- Lee C. Dose relationships of phase II, tachyphylaxis and train-of-four fade in suxamethonium-induced dual neuromuscular block in man. Br J Anaesth. 1975 Aug;47(8):841-5. doi: 10.1093/bja/47.8.841. PMID: 1201162.
Common questions
What is the mechanism of action of pancuronium?
Pancuronium is a competitive non-depolarising neuromuscular blocking agent that antagonises acetylcholine at the post-synaptic nicotinic N₂ receptor on skeletal muscle. It also raises heart rate, blood pressure, and cardiac output by two mechanisms: a vagolytic effect (M2 muscarinic receptor blockade at the sinoatrial node) and sympathetic stimulation (blockade of noradrenaline reuptake plus ganglionic stimulation).
What is the intubating dose of pancuronium?
The intubating dose is 0.1 mg/kg IV, with onset at approximately 2.5 minutes and a duration of at least 90 minutes. Its prolonged action suits long cases such as cardiac surgery but carries a risk of re-curarisation if reversed too early.
What is the difference between Type I and Type II neuromuscular block?
Type I (depolarising) block from suxamethonium shows no fade on train-of-four and no post-tetanic potentiation. Type II (Phase II) block develops with repeated suxamethonium doses and mimics a non-depolarising pattern with fade, post-tetanic potentiation, and partial reversibility with anticholinesterases.
How is pancuronium eliminated?
Pancuronium is primarily eliminated by the kidneys (approximately 40–50%, of which around 80% is unchanged drug), with the remainder undergoing hepatic deacetylation to 3-hydroxypancuronium (approximately 40% the potency of the parent drug). Renal failure significantly prolongs its duration of action, making it unsuitable for patients with impaired renal function. Its long clinical duration (90+ minutes) partly reflects this renal dependence.
What are the main side effects of pancuronium?
Vagolytic tachycardia, hypertension, and increased cardiac output, from M2 muscarinic blockade at the sinoatrial node together with sympathetic (noradrenaline-reuptake blockade and ganglionic) stimulation. Minimal histamine release. Prolonged duration of action is the chief clinical drawback. Potentiated by magnesium, gentamicin, volatile agents, and lithium.
Thanks for listening. Take it day by day, don't overcook yourself — keep studying.
Transcript
29 min listenRead the full transcript
GasGasGas EduCast: Pancuronium – The Original Aminosteroid
Introduction
[00:00–00:45]
Please listen carefully. Hello, Team Anaesthesia. Welcome to Gas Gas Gas. This is the best anaesthetic science podcast for the FRCA Primary exam. Our goal is to fill your brain with all this highly useful information. Now you might be in the gym right now, commuting, or ironing your scrubs, and there’s no judgement here. Gas Gas Gas will prime your brain for the monsoon of knowledge you need to imbibe. But regardless, the revision is eventually going to end, but for now, expect facts, concepts, model answers, and the odd tangent. Now remember to check out the website – that’s gasgasgas.uk. There are show notes there with all the detail, plus links to foundational reference papers and to anything else useful I find for you guys. Anyway, buckle up, get ready for your mind to be bent into a new shape, and let’s get on with the show.
Episode Opening – Why Pancuronium?
[00:46–01:32]
- Pancuronium is one of the earliest aminosteroid neuromuscular blocking agents to reach clinical use.
- Understanding pancuronium teaches us about the pharmacology of modern aminosteroids.
Hello and welcome to another episode of Gas Gas Gas. I hope you’ve had a delightful first two weeks of March. It’s finally something that could be mistaken for spring in deepest darkest Yorkshire. But let’s not get carried away with the sunshine – we’re here to talk about pancuronium, that exceedingly commonly used neuromuscular blocking agent which I’m sure is in everybody’s anaesthetic room fridge.
Why are we bothering to talk about it? Well, other than the fact that I just feel like I should, it is perhaps one of the first aminosteroid neuromuscular blocking agents that really made it to market. And we can learn a lot about today’s aminosteroids from this one.
Naming and Historical Development
[01:33–04:37]
- Name derived from “piperidino androstane curarising onium” – referencing its steroid hormone backbone.
- First synthesised in 1964 by Buckett, Hewett, and Savage; brand name: Pavulon.
- Development involved grafting acetylcholine-like moieties onto steroid hormone scaffolds.
- Bowman’s principle (Professor Bill Bowman, Strathclyde University): potency is inversely proportional to onset time.
So the ampoule will be labelled pancuronium bromide and its brand name once upon a time was Pavulon. Where does the name pancuronium come from? The name was derived from it being called a piperidino androstane curarising onium. Who knows why they put “onium” on the end? Curare. Androstane is an important word there. I’m sure once upon a time, at the deepest depths of studying steroids at medical school and testosterone and all that sort of stuff, you’ll remember the word androstenedione, and that is because this molecule is based on a steroid hormone.
Ultimately, the original attempts at aminosteroids involved putting an acetylcholine-like molecule onto either a 5-alpha-androstane-17-one or 5-alpha-pregnan-20-one – which is a synthetic androstenedione – and then later on to an endogenous steroid hormone. And when they were mucking around, they found that these fudged acetylcholine–steroid molecules had a degree of paralysing activity.
Now, at that point in time, someone called Buckett (with two T’s) noted that it had limited clinical value, but that these compounds presented leads for further chemical exploration, particularly the subsequent synthesis of bisquaternary aminosteroidal salts. Buckett, Hewett, and Savage in 1967 pointed out a bunch of these different molecules, one of them being pancuronium bromide, which they first synthesised in 1964. So this has been around since the sixties. This was an exciting time for anaesthetic pharmacological science. Nowadays I don’t think we quite do as much anaesthetic drug discovery. Everything we’ve got’s quite good – except for the odd thing here or there like sugammadex, a future episode.
In amongst all this joyful reading of papers that have been scanned because they were typed rather than printed once upon a time, I stumble across where Bowman’s principle actually derives from. Now, you remember Strathclyde University were heavily involved in anaesthetic science, particularly neuromuscular blockade, where they developed atracurium. So Professor Bill Bowman of Strathclyde University spent a lot of time fiddling around with neuromuscular blocking drugs, and this is where the term Bowman’s principle comes from, because he posited and demonstrated that the potency of a drug is inversely proportional to its onset time – because you have to give more of the less potent drug. Bowman’s principle. Bit more history there.
Pharmaceutical Properties
[04:38–05:06]
- Presented as a clear, colourless solution; must be refrigerated.
- Single stereoisomer; acidic solution; reportedly bitter taste.
- Molecular weight: 732 g/mol (heavier than rocuronium).
- Standard ampoule: 4 mg.
So back to pancuronium bromide. It is a single stereoisomer. It is presented as a clear and colourless solution that must be refrigerated, with four milligrams classically in an ampoule. It is acidic, apparently it has a bitter taste if you’re going around tasting paralysing drugs. And it’s quite a heavy molecule in the grand scheme of neuromuscular blocking drugs – it’s heavier than rocuronium at 732 grams per mole.
Mechanism of Action
[05:07–06:23]
- Competitive, non-depolarising neuromuscular blocking drug.
- Antagonises acetylcholine at post-synaptic N₂ nicotinic receptors on skeletal muscle.
- Also acts on pre-synaptic receptors, blocking retrograde modulation of acetylcholine mobilisation (the basis of fade).
- Vagolytic/ganglionic blocking effect at N₁ receptors → sympathetic predominance → increased heart rate, blood pressure, and cardiac output.
Mechanism of action of pancuronium – you’ve guessed it. It is a competitive, non-depolarising neuromuscular blocking drug. It works at the neuromuscular junction, antagonising the action of acetylcholine at the nicotinic receptor. Remember that’s the N₂ receptor situated on the post-synaptic membrane of the skeletal neuromuscular junction. But it also has action on the pre-synaptic membrane. Remember, this is the retrograde modulation of that motor neurone. By acetylcholine – when it’s released into the neuromuscular junction, some of that acetylcholine goes back and tickles that motor neuron ending to say, “Hey, I’ve just had to contract. You need to mobilise some more acetylcholine and get those vesicles nearer that synaptic cleft, ready to be released. If another signal comes, don’t be lazy, get to work.” It’s a bit of a feedback loop. This is where fade comes from.
And pancuronium does have an action in the ganglion blocking department. Ganglion – really we’re talking about the parasympathetic nervous system here, and it will bind to and impair the function of your parasympathetic nervous system. That means that you tip towards a bit more of a sympathetic drive, leading to an increase in heart rate, blood pressure, and cardiac output.
Dosing and Clinical Use
[06:24–07:21]
- Intubating dose: 0.1 mg/kg.
- Onset to intubating conditions: approximately 2.5 minutes; peak effect at ~4 minutes.
- Duration of action: at least 90 minutes (longer in cirrhotic patients).
- No maintenance infusion typically required – “fire and forget.”
- Favoured in long cases (e.g. cardiac surgery); unsuitable for short procedures.
- Risk of re-curarisation if reversed too early with neostigmine.
How do you dose it? If we think, well, there’s only four milligrams in a vial, and generally speaking a vial of drug is about one adult dose, here and there, depending on the drug of choice, of course. I wouldn’t really classify a four milligram ampoule of noradrenaline as one adult dose – that would probably be quite problematic. But four milligrams of ondansetron in an ampoule – certainly one adult dose.
So the intubating dose of pancuronium is 0.1 milligrams per kilo, taking two and a half minutes to get you to something that could be mistaken for intubating conditions, and peak effect is about four minutes. Do you need a maintenance infusion? Well, not really, because pancuronium lasts for a really quite long time – at least ninety minutes, give or take. If you’re a cirrhotic patient, even longer. So it was a bit of a fire and forget. They’re gonna be paralysed for an hour and a half. Put your feet up. Great for a long case. Not so great for a quick ENT procedure.
So the important thing to think about with a competitive non-depolarising neuromuscular blocking drug that lasts a really long time is that the degree of recovery takes a really long time. So instead of maybe seeing someone snap from a scant bare four twitches with fade to barely any fade, that is a longer journey. But if you’re seeing four twitches and you’re tempted to reverse the patient, you might find yourself in a situation whereby you’ve made it all better, the neostigmine wears off, and actually that pancuronium is still lurking, wanting to fight it out with the acetylcholine in the neuromuscular junction – leading to re-curarisation.
Oral Toxicology – A Historical Curiosity
[07:22–09:37]
- Lowest paralysing dose (non-human model): 0.01 mg/kg.
- Oral LD in mice: 21.9 mg/kg.
- Extrapolated lethal oral dose in an adult: ~1.5 g (approximately 400 ampoules).
- Rocuronium (with greater hepatic metabolism): extrapolated ~120 ampoules orally.
So the lowest paralysing dose in a non-human model is actually a tenth of the intubating dose recommended – so 0.01 milligrams per kilo. Now, the wonders of historic science have led me to an exciting discovery. You know I’ve taken the mickey several times saying, well, if you drink this drug, it probably doesn’t do anything. Well, they did indeed give mice pancuronium orally. Now, how much pancuronium per kilo do you have to give a mouse orally to paralyse it and thus doom it? We need to give it 21.9 milligrams per kilo. Now obviously a mouse is quite small and less than a kilo, but that is quite a lot more pancuronium for it to be orally absorbed and cause mischief.
So rocuronium is also an aminosteroid, although with a greater liver metabolism. So we could infer that a lethal dose of pancuronium in an adult might be 1.5 grams of pancuronium – which, you know, that’s like four hundred ampoules or something stupid. Someone would probably notice, and there probably isn’t enough in the hospital. It might, if you’d fudge the maths and not consider hepatic metabolism of rocuronium, mean that you might need to consume perhaps 120 ampoules of rocuronium to paralyse yourself orally. Now I’ve not tasted rocuronium. I don’t recommend you should either, but there you go. Maybe they do have some oral effect after all.
Anyway, that was very exciting for me. You’re probably sat there thinking, James, I’m trying to pass this bloody exam – stop talking about eating rocuronium and pancuronium. You’re right, let’s move on.
Side Effects of Pancuronium
[09:38–10:47]
- Generally haemodynamically stable (unlike suxamethonium).
- Increased heart rate, blood pressure, and cardiac output (vagolytic effect via N₁ ganglionic binding).
- Very rarely causes histamine release or bronchospasm.
- Does not significantly alter systemic vascular resistance.
- Obliterates rate and depth of breathing (paralysis).
- Potentiated by co-administration of magnesium, gentamicin, volatile anaesthetic agents, lithium, etc.
- Chief clinical drawback: prolonged duration of action.
- Agent of choice in cardiac anaesthesia for long cases.
So, side effects of pancuronium. Like most other neuromuscular blocking drugs, except for suxamethonium, it’s pretty haemodynamically stable. It causes an increase in heart rate and blood pressure, and therefore a bumped cardiac output, due to this vagolytic effect – ganglionic effects where it goes off and binds to N₁ receptors, not N₂ receptors. Very unusual for it to cause histamine release, therefore very unusual for us to see bronchospasm with it. It doesn’t muck with the systemic vascular resistance particularly, unless you’re very unfortunate to see some histamine release. And naturally, it obliterates the rate and depth of breathing because it paralyses you.
As with other neuromuscular blocking agents, there are conditions which increase the potency of these drugs, such as co-administration of magnesium, gentamicin, volatile anaesthetic agents, lithium, etcetera – covered in a previous episode. The real chief problem with pancuronium is it hangs around for ages. But it was a choice agent in cardiac anaesthesia where you might be doing a six or seven hour case every day, and therefore why not make your life easier by just giving them some pancuronium and having a nicely paralysed patient for at least an hour and a half?
Pharmacokinetics
[10:47–12:58]
- Absorption: not applicable (IV administration); oral paralysis demonstrated only in murine models at very high doses.
- Distribution: Vd = 0.2 L/kg; chiefly water-soluble; increased Vd in cirrhotic patients.
- Metabolism: 30–45% hepatic metabolism by deacetylation; excreted via the biliary system.
- Active metabolite: 3-hydroxypancuronium (25% of metabolised drug), with 50% of parent compound activity.
- Excretion: 40–50% excreted unchanged in urine; ~80% of urinary content is unchanged pancuronium.
- Elimination half-life: 69–160 minutes (doubled in cirrhotic patients).
- Clearance: 1.1–2.2 mL/kg/min.
So, pancuronium pharmacokinetics. Absorption – I’ve written not applicable, because it isn’t really. But we know that you could achieve some paralysis with oral pancuronium. The onset time is probably not terribly compatible with intubation though.
Distribution. So you would expect, like rocuronium (which is another aminosteroid) or like atracurium (a benzylisoquinolinium), its volume of distribution is quite low – it is 0.2 litres per kilo. It’s chiefly water-soluble, doesn’t really soak into those fat stores. Its volume of distribution is higher in a cirrhotic patient. And bear in mind that these were cirrhotic patients from the 1960s and 70s, whereby our treatment and mentalities were perhaps not so great at managing the water content of someone with less than effective liver function, but ultimately they required a higher dose to achieve intubating conditions within a sensible period of time.
Metabolism. Like other aminosteroid drugs, it undergoes a degree of liver metabolism, although not quite a lot. 30 to 45% of the molecule is metabolised by deacetylation, and then it’s excreted in a manner through the biliary system. Now, one of the metabolites – a quarter of it – is converted into 3-hydroxypancuronium, and that has 50% of the activity of pancuronium. So we’ve got an active metabolite. But we now should be thinking, well, only thirty to forty-five percent is cleared by the liver. So actually forty to fifty percent is excreted in the urine, and about eighty percent of the content that ends up in the urine is unchanged – so it’s still pancuronium. So you whiz out about half to forty percent of the molecule unchanged.
Half-life: 69 to 160 minutes. And double it if you’ve got a cirrhotic patient. So if you are somehow in a situation where you’re doing an extensive coronary artery bypass or some other cardiac mischief in a cirrhotic patient – which you probably wouldn’t do – that dose of pancuronium might start to last you the whole case. And its clearance is 1.1 to 2.2 mL per kilo per minute. Exciting.
So, pancuronium. That was a whistle-stop tour.
[Sponsor Break – Teach Me Anaesthetics]
[13:02–14:30]
Sponsor segment: Teach Me Anaesthetics – an SBA question resource for the FRCA Primary exam with 1,100+ questions, detailed explainers, and multiple study modes. Affiliate links available via gasgasgas.uk.
Type I and Type II Neuromuscular Block
[14:31–16:05]
- Type I block: associated with depolarising agents (suxamethonium); no fade on train-of-four.
- Type II block: associated with non-depolarising agents; demonstrates fade, post-tetanic potentiation.
- Also called a desensitisation or dual block.
- Repeated doses of suxamethonium can shift a Type I block to a Type II block.
How much more can we possibly talk about neuromuscular blocking drugs? Well, seeing as they’re very commonly used – plenty. I’m sure you’ve come across, someone’s mentioned, or you’ve seen in some esoteric literature a mention of Type I and Type II neuromuscular block. Now this really did confuse the bejesus out of me, because I hadn’t heard that expression before. It’s important that we just cover it so that if someone mentions it, or it perhaps comes up in an exam when someone’s using a slightly awkward term to test you.
And it refers to the behaviour of the neuromuscular junction when tested with a nerve stimulator, dependent on the class of drug it’s exposed to. So a Type I block is a block associated with the administration of suxamethonium – you’ve tested that neuromuscular junction with the nerve stimulator when you’ve given a dose of suxamethonium. And a Type II block, sometimes described as a desensitisation or dual block – another thing to confuse the heck out of you – is generally associated with non-depolarising neuromuscular blocking drugs. But it would also be pertinent to say that a Type II block demonstrates fade, whereas a Type I block does not demonstrate fade. That’s perhaps one of the key differentiators. The other foible, interesting or not, is that if you give someone repeated doses of suxamethonium, you shift from a Type I block to a Type II block. And we’re going to explore that too.
Type I (Depolarising) Block in Detail
[16:06–20:12]
- Suxamethonium acts as an agonist at post-junctional N₂ receptors, causing depolarisation.
- Fasciculations arise from disordered depolarisation across the muscle as suxamethonium reaches different motor endplates at slightly different times.
- Acetylcholinesterase in the neuromuscular junction cannot metabolise suxamethonium; it must diffuse back to plasma for clearance by plasma cholinesterase (butyrylcholinesterase).
- No fade on train-of-four: each stimulus produces the same diminished amplitude because suxamethonium binding is non-competitive.
- No post-tetanic potentiation.
Now, I don’t know how many people have attached their nerve stimulator to a suxamethonium-anaesthetised patient, but I have. Mainly because I was just wondering – when do they get their twitches back? Are they in that cohort of people who are paralysed for a little bit longer from suxamethonium? Just for curiosity. I can’t tell you I found anyone who was a bit more paralysed than you would otherwise expect. But when you do twitch them, you won’t see fade. You’ll just see a diminished response and a similar amplitude of contraction with each stimulus delivery.
Type I depolarising block. Suxamethonium is the only drug we use in this category, and we know that suxamethonium binds to the post-junctional nicotinic acetylcholine receptors and acts as an agonist – i.e. it activates those N₂ receptors, causing depolarisation. This depolarisation leads to fasciculations because that depolarisation is slightly disordered. It’s not all the muscle contracting all at once. It’s all the bits of the muscle that have achieved a suxamethonium concentration at slightly varying times because of blood flow across the muscle, leading to fascicles depolarising at different times – and you get that twitchy phase, and then paralysis. So it does give you a lovely endpoint of when they’re paralysed, and it does work really quickly – faster than rocuronium, really.
However, it is not broken down in this cleft – in this neuromuscular junction – because acetylcholinesterase there can’t touch it. Therefore suxamethonium floats around in there until it is cleared back to the plasma. Now, you are right in remembering that quite a bit of a dose of suxamethonium is cleared in the plasma before it actually makes it to the neuromuscular junction. But once it’s soaked into the neuromuscular junction, the drug that is there works. It has to diffuse back out for it to be cleared. This is because plasma cholinesterases can knock it on the head, whereas acetylcholinesterase in the neuromuscular junction cannot.
So we’re in a situation where we’ve got a neuromuscular junction flooded with suxamethonium that’s falling off receptors, rebinding to receptors, and this keeps the resting membrane potential in an awkward situation whereby it cannot repolarise and be ready for the next signal. Some acetylcholine’s binding, some of it’s floating off, some’s binding to something else, until it eventually clears enough that it can repolarise.
As this suxamethonium eventually clears out, you find yourselves in a position where you can trigger a bit of a contraction in the muscle, but not a lot. This is because some of our nicotinic acetylcholine-receptor sodium channels are blocked and some are not. Therefore, if you can deposit some acetylcholine into the neuromuscular junction, some of it will make it across, some of it will bind to these now accessible receptors and trigger a degree of contraction. However, the amount of acetylcholine in the neuromuscular junction released by an action potential down that motor neurone – that amount of acetylcholine is the same, but it’s working against a barrier of suxamethonium, so to speak, so it will only achieve the same effect each time. No amount of increased acetylcholine in the neuromuscular junction will increase the amount of muscle you can trigger a contraction in, because it’s not a competitive situation – that suxamethonium is bound, and it won’t unbind just because there’s some acetylcholine knocking around.
So, unlike a Type II block (or a Phase II block, or a dual block, or a desensitisation block – all the different names for the same thing), you won’t see fade. And even if you do a tetanic stimulus and then do a post-tetanic count, you won’t see fade. You’ll just see the same meek contraction each time, but it will get better – you’ll get more of a contraction as the suxamethonium slowly clears out.
Type II (Phase II / Desensitisation) Block in Detail
[20:13–25:06]
- Resembles a non-depolarising block despite being caused by a depolarising drug.
- Demonstrates fade on train-of-four, tetanic fade, and post-tetanic potentiation.
- The nicotinic receptor transitions from an open (depolarised) state to a desensitised (closed) state with suxamethonium still bound.
- Na⁺/K⁺-ATPases gradually restore membrane potential, but closed receptors remain unresponsive.
- Standard dose (1–2 mg/kg) remains in Phase I; repeated/cumulative doses (3–5 mg/kg) shift to Phase II.
- Tachyphylaxis develops: progressively more suxamethonium needed.
- Giving rocuronium to a patient in Phase II block deepens paralysis.
- Anticholinesterases may partially reverse a Phase II block (increased acetylcholine outcompetes at open receptors).
- Neostigmine given during a Phase I block prolongs the depolarising blockade.
A Type II block. This is different, as you would expect to Type I, and resembles the block produced by non-depolarising agents, despite this block being caused by a depolarising drug. And it might make your head hurt a little bit.
So what do you see? You see fade across the train-of-four, tetanic fade, and post-tetanic potentiation – that is, you fire off a train-of-four and then immediately fire off another train-of-four; the second one you will probably see an increased amplitude of contraction compared to the first. You have potentiated the neuromuscular junction. This is what you see with non-depolarising competitive neuromuscular blocking drugs like atracurium, rocuronium, mivacurium, etcetera.
I’m not gonna come out and tell you we know exactly why this happens, because it doesn’t appear that we do. But actually, if you drill down into most things in medicine, we’ve got a bit of a grip of it, but when you start getting into the edge cases and looking at the minutiae, you think, hmm, there’s something here that we’re not quite grasping. So don’t stress about it too much.
The crux of what we do know is that the nicotinic receptor that the suxamethonium is tickling transitions from being depolarised – i.e. open, allowing sodium through it, depolarising the membrane – to a desensitised state. It’s hung up its coat, it’s had enough. It’s closed, but occupied by the suxamethonium. So it becomes just unresponsive to further stimulation, because it’s closed. Those sodium–potassium ATPases that are very busy little fellows can start getting that muscle creeping back towards a repolarised state.
So as some of that suxamethonium clears out of the junction, but some of it is just bound to receptors that aren’t going to do anything, you find yourself in a situation where acetylcholine delivered to the neuromuscular junction by an action potential down a motor neurone can trigger a contraction.
Now, why does it cause fade? I don’t think anyone’s particularly clear on that. But what is important to know is that your standard one to two, and maybe up to three, milligrams per kilo dose of suxamethonium will keep you in Phase I block land. Whereas if you’ve given repeat doses – three to five milligrams per kilo – you creep into this Phase II block land.
What are the issues with this Phase II block? Well, it’s unpredictable. It lasts quite a long time. If you’re trying to maintain paralysis with suxamethonium, you see a degree of tachyphylaxis – because remember, these receptors have shut up shop and they’re not bothered. So you need more and more suxamethonium to try and bind those acetylcholine receptors that are still active. So it becomes quite unclear as to where you are and whether you can actually maintain paralysis.
And as you might expect, we see suxamethonium apnoea in patients. We’ve given them one to two milligrams per kilo. If you’ve got someone who has reduced plasma cholinesterase activity – either because they’re in that small group of people where it lasts a little bit longer, or pregnant patients or those with liver disease who aren’t producing as much plasma cholinesterase – there you might find yourself in a world where your suxamethonium dose number two lasts a really long time. One of the reasons why suxamethonium infusions for paralysis are not modern practice at all.
So, can we do anything about how long it lasts? Can we give any drugs to fix this problem? If you were – first things first – what happens if you give rocuronium to someone who’s in a Phase II block? Well, you’ve just paralysed them more, because those receptors that were amenable to being blocked now have a competitive blockade courtesy of the rocuronium.
What happens if you give an anticholinesterase to someone in a Phase II block? You might see a degree of reversal, as you’ve bumped up their acetylcholine concentration in the neuromuscular junction. So it might push you away from being in a Phase II block. But if you were to give neostigmine to someone who you’d just given suxamethonium to, you would actually extend the effect of that blockade. Because you’ve now introduced more acetylcholine into the neuromuscular junction with the suxamethonium floating around in it – it’s going to bind to the odd receptor that the suxamethonium is not bound to, depolarising that receptor or causing it to open, allowing sodium in. So neostigmine will make suxamethonium work longer in a Phase I block. It all gets a bit spooky and mischievous. And this is why everyone just reaches for rocuronium these days.
Side Effects of Suxamethonium
[25:06–26:28]
- Malignant hyperthermia.
- Suxamethonium apnoea.
- Anaphylaxis.
- Significant bronchospasm.
- Myalgia.
- Life-threatening hyperkalaemia in patients with denervation injuries, muscular dystrophy, significant cerebral palsy, or burns >48 hours old (due to extrajunctional nicotinic receptor upregulation).
- Raised intraocular pressure and intracranial pressure.
- Bradycardia with repeat doses (especially in children).
Suxamethonium – loads of side effects. The only benefit is it is very fast in onset and pretty fast in offset. But if they ask you in the exam, “What are the side effects of suxamethonium, Doctor?” – you need to be able to list a reasonable number of them.
Bad side effects of suxamethonium: malignant hyperthermia – bad news. Suxamethonium apnoea – also problematic. And anaphylaxis – very challenging. Less problematic but still could give you the heebie-jeebies: it randomly causes significant bronchospasm that might mean you feel like you can’t ventilate the patient and think you’re in the oesophagus. It causes significant myalgia and muscle aches. In patients who are poorly innervated in their musculature – courtesy of muscular dystrophy or significant cerebral palsy, or those who’ve been burnt more than forty-eight hours ago with increased extrajunctional nicotinic receptors – you will cause a life-threatening hyperkalaemia when you give suxamethonium. It can raise intraocular pressure and intracranial pressure. Repeat doses cause bradycardia, especially in children. Generally, all around, not a great drug.
Summary
[26:28–27:40]
- Phase I block: straightforward depolarising block; diminished but equal-amplitude twitches; no fade.
- Phase II block: develops with prolonged/repeated suxamethonium; less predictable; demonstrates fade and post-tetanic potentiation.
- Phase I → Phase II transition is dose-dependent and coincides with tachyphylaxis.
- Phase II blocks are partially reversible with anticholinesterases; Phase I blocks are potentiated by anticholinesterases.
- Pathophysiology of Phase II block: desensitisation of the nicotinic receptor into a closed state with suxamethonium still bound.
So remember, in summary: a Phase I block is that straightforward depolarised block where you might see a twitch, but it’s just gonna be the same amplitude every time as that block recovers. Phase II block develops with prolonged or repeat suxamethonium doses, and I think it’s important to make a distinction that it’s not really the same sort of block as that purely driven by non-depolarising neuromuscular blockade, because it’s less predictable, causes weird mischief, and behaves in a strange manner. Differentiate them in your mind. Phase I and Phase II really pertain to how suxamethonium behaves.
That transition from Phase I to Phase II is dose-dependent, can happen quite suddenly, and coincides with tachyphylaxis – i.e. they become less responsive to the paralysing effects of suxamethonium. Phase II blocks can be partially antagonised with anticholinesterases, whereas Phase I blocks are potentiated by cholinesterases. And if in doubt and anyone’s really trying to drill you down on the pathophysiological basis of a Phase II block, say: no one really 100% knows, but we know that it desensitises the nicotinic receptor into a closed state with a suxamethonium molecule still bound to it.
Outro
[27:40–29:07]
How exciting – pancuronium. That’s done. It was pandemonium with pancuronium. I should have probably got that in at the start. But you’ve gone, you’ve done it, you’ve listened. Raw flabbergasted that 21.9 milligrams per kilo of oral pancuronium can indeed paralyse a mouse. But we’re not gonna go out and do that. But at least we’ve learnt something vitally important. I hope you enjoy the next two weeks pondering such mysteries. And next we’re gonna do reversal – so that’s neostigmine, glycopyrrolate, and sugammadex.
You’ve been listening to Gas Gas Gas. This was James Waffling On. Have a lovely weekend. Cheerio and goodbye.
Ahoy, Team Anaesthesia. You’ve survived yet another episode of Gas Gas Gas. Now if you’ve found it useful or harrowingly awful, please like and subscribe, drop us a star or twelve, and follow with whichever podcast platform you find yourself using. Please leave a comment or ping off an email if you think I need to square something away. Now there are a bunch of ways to support the costs of Gas Gas Gas – from buying me a coffee to venturing forth via an affiliate link to the horde of joyful SBA questions from Teach Me Anaesthetics. Those links are on the website and in the show notes. Speaking of the website – definitely check out gasgasgas.uk for the show notes, diagrams, the details, the references. Now we all know, guys, that this is a bucket of content to consume, and this is like drinking from a fire hose. So I want to finish by saying: take it day by day, don’t overcook yourself, don’t freak out, and keep studying.
Enjoyed this? Review on Apple Podcasts Rate on Spotify
Support the show Help keep the lights on SBA question bank @ Teach Me Anaesthetics